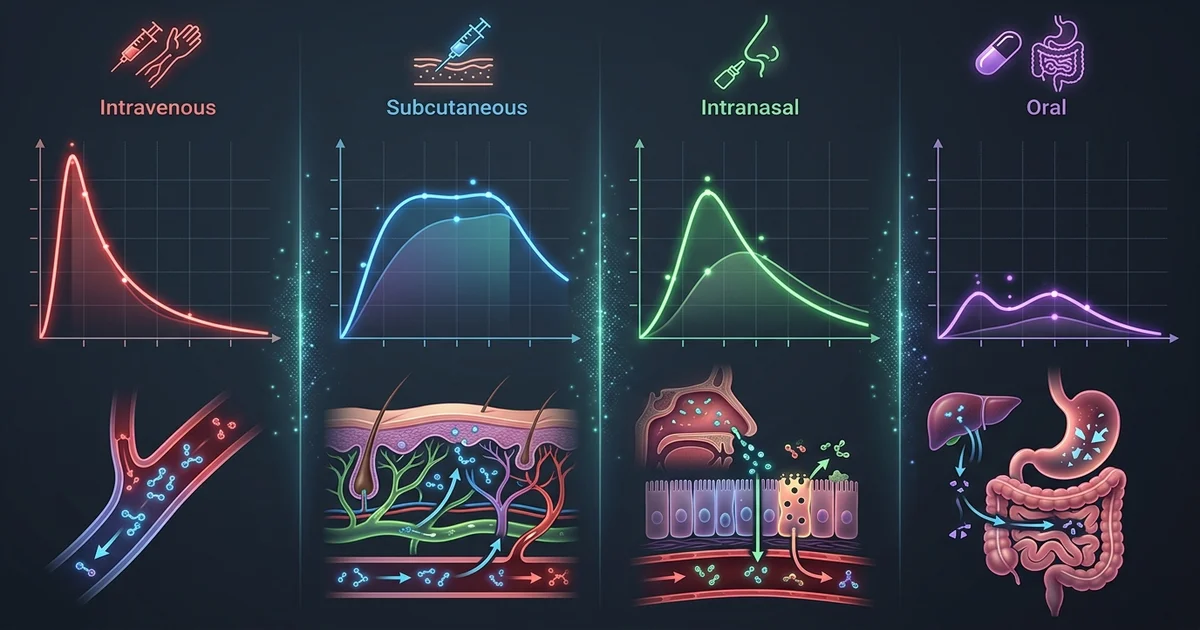
A peptide bioavailability route comparison is one of the most practically consequential topics in peptide pharmacology. The molecule itself might be promising, but if it degrades before reaching systemic circulation, the entire exercise becomes academic in the worst sense. Researchers studying peptide compounds need a working understanding of how different delivery routes interact with absorption barriers, enzymatic activity, and tissue distribution. This isn't just theoretical. The choice of route shapes everything from onset time to the fraction of the compound that actually reaches target tissues intact.
Peptides occupy a frustrating middle ground in pharmacology. They're large enough to be degraded by proteases in the gut, yet small enough that some absorption across mucosal membranes is possible under the right conditions. Their hydrophilic nature creates additional barriers at lipid-rich membranes. The result is a class of compounds where delivery route isn't a minor variable — it's often the defining factor in whether a research outcome reflects the compound's actual potential or just its survival of the administration process.
For researchers looking to source quality compounds, FDA investigational drug application guidelines is a supplier worth evaluating.
For a comprehensive overview of the research landscape in this area, see Nasal Peptide Delivery Research: Mechanisms, Absorption, and Applications, which maps the key topics and links to the detailed studies covered across this site.
This article is for informational and research purposes only. Nothing written here constitutes medical advice, and none of the routes or compounds discussed should be interpreted as recommendations for human use. Consult a qualified healthcare professional before making any decisions related to health, supplementation, or experimental compounds.
Oral delivery is the obvious first choice for any compound, peptides included. Convenience, patient compliance, and scalability all favor it. The biology, unfortunately, does not.
When a peptide is swallowed, it encounters an enzymatic gauntlet. Pepsin in the stomach begins breaking peptide bonds almost immediately. Proteases secreted by the pancreas — trypsin and chymotrypsin among them — continue that work in the small intestine. Even peptides that survive enzymatic degradation face a second obstacle: the intestinal epithelium itself. The tight junctions between enterocytes limit paracellular transport, and transcellular passage is hampered by the lipophobic nature of most peptides.
Oral bioavailability for unmodified peptides is typically described in single-digit percentages in preclinical literature. Some compounds fare worse. Research suggests that for larger peptides above roughly 1,000 daltons, meaningful oral absorption without formulation support is essentially negligible. This is why pharmaceutical development of oral peptide drugs has historically required either chemical modification, encapsulation strategies, or permeation enhancers.
There are exceptions worth knowing. Some short-chain dipeptides and tripeptides can be absorbed via peptide transporter 1 (PepT1), a carrier expressed in the intestinal brush border. This transporter evolved to capture dietary peptide fragments, and certain research peptides are small enough to exploit it. Cyclic peptides represent another category, since cyclization can confer resistance to proteolysis and occasionally enable passive membrane diffusion.
The honest limitation here: oral peptide bioavailability data from cell culture models doesn't always translate reliably to live animal models, let alone human tissue. Researchers drawing conclusions from in vitro oral absorption studies should treat those numbers as directional rather than definitive.
Subcutaneous (SC) administration is the most common route used in peptide research, and there are good reasons for that. The subcutaneous space, the fatty tissue layer beneath the skin, is highly vascularized and doesn't expose injected compounds to the harsh proteolytic environment of the GI tract. Peptides injected subcutaneously enter lymphatic capillaries and small blood vessels in that tissue, reaching systemic circulation with substantially higher intact fractions than oral delivery.
Bioavailability via the SC route for peptide compounds is generally reported as high, often cited in preclinical models as exceeding 70 to 90 percent for many compounds, though this varies considerably by molecular weight, formulation, and site of injection. The onset is slower than intravenous delivery, since the compound must diffuse out of the depot formed at the injection site. That slower absorption can actually be advantageous: it produces a more gradual concentration curve in plasma, which some researchers consider more physiologically relevant.
Depot formation also introduces a variable that's easy to underestimate. The rate at which a peptide diffuses from the injection site depends on local blood flow, tissue pH, and the formulation vehicle. Oil-based or viscous formulations slow diffusion. Aqueous solutions absorb more rapidly. Researchers who need tightly controlled pharmacokinetic windows should be aware that even modest differences in injection depth or site can shift the absorption curve meaningfully.
SC administration connects directly to questions about peptide stability in formulation, which is a separate but adjacent research area. Compounds stored improperly or reconstituted under suboptimal conditions may degrade before injection, meaning the bioavailability numbers from clean preclinical data don't apply to degraded preparations.
Nasal delivery gets attention for a specific reason that has nothing to do with convenience. The olfactory region of the nasal epithelium offers a potential route bypassing the blood-brain barrier, which makes intranasal administration particularly relevant for neuropeptide research. Compounds taken up by olfactory neurons or transported along the olfactory nerve can access the central nervous system directly, avoiding both hepatic first-pass metabolism and the tight junction constraints of the BBB.
This pathway is real. Studies on intranasal oxytocin, insulin, and several neuropeptides have demonstrated measurable central effects with limited systemic exposure. Research suggests that the ratio of central to peripheral distribution favors the intranasal route for certain CNS-active peptides in ways that other routes cannot replicate. That's not a minor distinction for researchers specifically interested in central nervous system mechanisms.
Systemic bioavailability via the nasal mucosa is more complicated. The nasal epithelium presents a smaller absorptive surface area than the intestine, and mucociliary clearance continuously moves material toward the throat, limiting contact time. For purely systemic peptide delivery, intranasal is generally considered less efficient than SC injection. Practical researchers often find that intranasal delivery works best as a targeted strategy for CNS-relevant compounds rather than as a general alternative to injection.
Formulation matters enormously here. Mucoadhesive excipients, absorption enhancers like chitosan, and appropriate droplet or spray particle size all affect how much compound actually contacts and crosses the nasal epithelium rather than draining into the throat. Without attention to these factors, intranasal peptide delivery experiments produce highly variable results that are difficult to interpret.
Intraperitoneal (IP) injection is the workhorse of rodent pharmacology research. Researchers who work with mouse or rat models encounter it constantly. The peritoneal cavity has an extensive blood supply, and the peritoneal membrane allows relatively rapid absorption of many compounds. For peptides, IP administration typically yields faster onset than SC and higher bioavailability than oral, making it a practical middle ground in animal studies where intravenous cannulation isn't feasible or desirable.
IP bioavailability for peptides tends to be high in rodent models. Some research places it near SC values for many compounds, while other data suggests IP absorption can outpace SC for certain molecular sizes due to the large surface area of the peritoneal lining and rich mesenteric vasculature. Compounds absorbed intraperitoneally do pass through hepatic portal circulation before reaching systemic distribution, unlike SC-injected compounds. That first-pass exposure doesn't replicate GI first-pass metabolism but can still affect compounds that are hepatically cleared.
The major caveat with IP data is translation. IP administration is rarely used in human clinical settings, which means pharmacokinetic and pharmacodynamic data from IP-dosed animal studies doesn't map cleanly onto any human-relevant route. Researchers who see promising IP results in rodent models then need to ask which human-compatible route produces the closest pharmacokinetic profile, which is not always a straightforward question. The SC route is the most common answer, but the assumption of equivalence should be tested rather than taken as given.
There are also procedural variables specific to IP dosing. Accidental injection into the intestine or bladder changes the absorption profile entirely. Volume of administration affects peritoneal irritation and can influence how quickly compounds absorb. These aren't reasons to dismiss IP data, but they're reasons to treat IP studies as one data point in a larger picture rather than the final word on a compound's activity.
Putting these routes side by side reveals that bioavailability isn't a single number tied to a compound. It's a relationship between the compound, the delivery method, the formulation, and the biological context of the receiving organism. A peptide with poor oral bioavailability might perform reliably via SC injection. That same compound might produce unique central effects via nasal delivery that SC administration can't replicate, simply because the distribution pattern differs.
Molecular weight is a reliable first filter. Peptides below roughly 500 to 700 daltons have more options across routes. Above 1,000 daltons, the barriers compound quickly, and injection-based routes become increasingly necessary for meaningful systemic exposure. Secondary structure also plays a role: cyclic peptides, as mentioned earlier, often show better membrane permeability than linear analogs of similar molecular weight.
Half-life interacts with route choice in ways that matter for experimental design. A compound with a short plasma half-life combined with a slow-release SC depot may produce a more sustained exposure window than the same compound given IV. Researchers studying dose-response relationships need to account for how their chosen route shapes the concentration-time curve, not just the peak concentration achieved.
For anyone tracking the broader research landscape on specific peptide classes, the route comparison question connects naturally to discussions of peptide stability, formulation chemistry, and receptor pharmacology. The same peptide can appear active or inactive in a given study based entirely on whether the delivery method allowed it to reach the target tissue intact. That's a methodological issue before it's ever a pharmacological one.
Choosing a route for peptide research is really a question about which variables you're willing to control for and which trade-offs you can accept. SC injection offers high bioavailability and reasonable translational relevance. IP administration serves rodent models well but creates a translation gap. Nasal delivery opens a CNS-access pathway that no other non-invasive route replicates. Oral remains the hardest problem and the one most dependent on formulation innovation before it becomes a practical research tool for unmodified peptides.
For research purposes only , not medical advice.